编者按:
Noster & Science 微生物组学奖由 Noster 公司和 Science 杂志共同设立。
Noster 公司于 2020 年 5 月 15 日在日本东京成立,专注于研发以肠道微生物组为基础的治疗方法,其愿景是“将生命与肠道微生物组相连接”。公司的使命是阐明 1000 种与人类共生的肠道微生物,为微生物科学的进步作出贡献,以改善全世界人民的健康。
近期,Science 杂志公布了该奖项的获奖结果,表彰并奖励了 3 位在微生物组学方面做出创新研究的年轻科研人员。其中,我们欣喜地看到一位中国学者出现在了获奖名单中。
今天,我们共同关注 3 位获奖者以及他们的研究成果。希望本文能够为相关的产业人士和诸位读者带来一些启发与帮助。
第一位获奖者Chun-Jun (CJ) Guo
Chun-Jun(CJ)Guo 获得了复旦大学的学士学位和南加州大学的博士学位。在加州大学旧金山分校和斯坦福大学 Fischbach 组完成博士后研究后,于 2018 年在康乃尔医学院的 Jill Roberts 研究所开设了自己的实验室。
CJ 的研究通过基于 CRISPR 的微生物遗传学、最先进的分析化学以及健康和疾病老鼠模型来探究微生物的基因、通路和代谢物如何影响宿主生物学。
以下是他的研究成果介绍:
数十年来使用各种组学方法探究微生物组的研究已经揭示了微生物组与人体健康之间的密切联系。下一步就是确定造成这种关联背后的分子作用机制:比如哪些菌群、基因和它们的代谢物造就了宿主的表型?如果这些物质确实能影响我们,我们要怎样调节它们在宿主中的比例来促进肠道健康?
从分子机制的水平揭露肠道菌群与宿主生物学之间的因果关系极具挑战,这主要是因为我们缺乏一种有效的方式来操控肠道微生物的基因。
现在,通过宏基因组测序已经发现了超过 90,000 种肠道微生物1,并且分离出了上千种细菌,并对它们的基因进行了测序,但是我们只能对其中不到 5%的菌种进行基因操作。要研究这些非模式生物的肠道微生物,迫切需要新的工具帮助。
肠道菌群通过各种方式调节宿主生物学。实现这种调节的主要手段之一是微生物的代谢产物。就像内分泌腺释放荷尔蒙,这些物质有三个明显的特征:它们只能由肠道微生物产生释放;其中一些代谢物的数量相当多且能进入血液循环;另一些则是宿主受体的配体。
CJ 的研究集中于探索微生物组代谢物与人类健康联系背后的分子机制。他的研究兴趣是开发新的基因工具来分离人体肠道中的梭菌、控制宿主中微生物组基因的表达和代谢物的含量并研究它们对于人体健康和疾病的影响。
CJ 的长期目标是理解宿主-微生物组之间联系的分子“语言”,并最终重新“组织”用于医疗。
之前针对宿主中微生物来源的分子的研究主要使用了 2 种方法:通过注射或灌胃引入一种化合物,或是调节产生目标代谢物的菌群组成。两种方法都使我们了解到生物功能的机制,但是缺乏一个清晰的背景或对照(如目标代谢物的生理学浓度,或是引入的微生物的其他生理活动)。
在 CJ 的实验室,他们认为研究宿主中微生物来源的代谢物最精准的方式,就是比较两种只在某个代谢物上有差别的微生物。要进行这样的试验需要确定负责产生这种代谢物的基因,以及一套用于操控肠道微生物基因的遗传工具。
目前,应用这一方法存在一个关键技术障碍:许多含量丰富的肠道微生物代谢物是由梭菌及其近亲产生的,而这些细菌是众所周知的难以进行遗传操作。
在 CJ 的工作中,他们在一种来源于肠道的梭状芽孢杆菌中引入了 CRISPR-Cas9 系统。梭状芽孢杆菌是一种常见于健康人体内的肠道共生菌,其发酵产生的许多物质能够进入血液循环。
尽管在其他许多细胞中也应用过 CRISPR-Cas9 系统,比如哺乳动物细胞和大肠杆菌(已了解其基因组的),但是在梭菌中建立这样一个系统并非易事。
梭菌属,之所以难以进行遗传操作,主要是因为没有有效的方法可以引入胞外的 DNA,而且大部分梭菌内源性同源重组频率很低,这也进一步限制了 CRISPR-Cas9 系统在其中的应用可行性。
历经超过 9 个月的努力,最终 CJ 发现向导 DNA 和 Cas9 组分分别引入梭状芽孢杆菌中能够有效促进的胞外 DNA 吸收和同源重组。
这一发现帮助 CJ 的团队找出并改变了 10 余个梭状芽胞发酵代谢物相关的基因。然后,通过给无菌小鼠接入基因编辑后的梭状芽孢杆菌和原始菌株,使得他们能够改变这些代谢物在宿主中的含量。
尽管像丙酸盐和丁酸盐这样的微生物代谢物是宿主 GPCR(G 蛋白结合受体)的配体,能够进入肠道中通过调节 T 细胞来调节免疫功能3,但是对于结构相似的支链短链脂肪酸(BSCFA)和它们的生物学机制几乎不为人知。
而 CJ 开发的这个系统使他们能够通过比较两组小鼠(两组小鼠仅在 BSCFAs 含量和相关代谢基因上有所差别)的差异,来了解 BSCFAs 的生物学功能。
他们发现,BSCFAs 降低了血浆中肠道免疫球蛋白 IgA 和许多先天免疫细胞表面的 IgA 浓度,这表明 BSCFAs 在调节 IgA 相关免疫细胞生物学上具有一定的作用,这是先前未被发现的。
一个健康人的肠道中含有数万亿微生物,但是只有 5%能够进行遗传操作。CJ 的团队计划建立一个通用的工作流程,促进针对这些先前无法进行基因操控的肠道微生物的基因工具的开发。
鉴于许多微生物调节的生物学表型需要复杂的微生物组定殖,他们的下一步目标是,组装一个遗传上可操控且兼具多样性的肠道微生物组。若能成功,这一系统将大大加快微生物与人相互作用背后的因果分子机制的阐明,也将为利用肠道微生物组促进人类肠道健康并防治疾病奠定基础。
第二位获奖者Mariana X. Byndloss
Mariana X. Byndloss 从巴西米纳斯吉拉斯联邦大学获得了兽医学博士。在加州大学戴维斯分校完成博士后研究后,Mariana 于 2018 年在范德比尔特大学医学中心的病理学、微生物学和免疫学系建立了她的实验室。
她的研究旨在了解炎症介导的肠道上皮代谢变化如何导致肠道菌群失调,并增加鼠伤寒沙门氏菌感染性胃肠炎和非传染性疾病(即肥胖相关心血管疾病和结肠癌)的风险。
以下是她的研究成果介绍:
微生物组是一个复杂的微生物生态系统,人类结肠是微生物的栖息地之一,结肠中的微生物组主要由厌氧微生物组成。最近的数据表明,肠道微生物及其代谢物可以通过多种机制影响人类健康,包括改变免疫反应 [1]、改变宿主细胞代谢状态[2],甚至影响对免疫疗法的反应[3]。
肠道菌群对健康和疾病的潜在关系是过去十年中最不寻常的发现之一。然而,我们才刚刚开始了解微生物促进肠道生理变化的机制,以及宿主和微生物组之间共生关系的变化对感染性和非感染性疾病发病机制的影响。
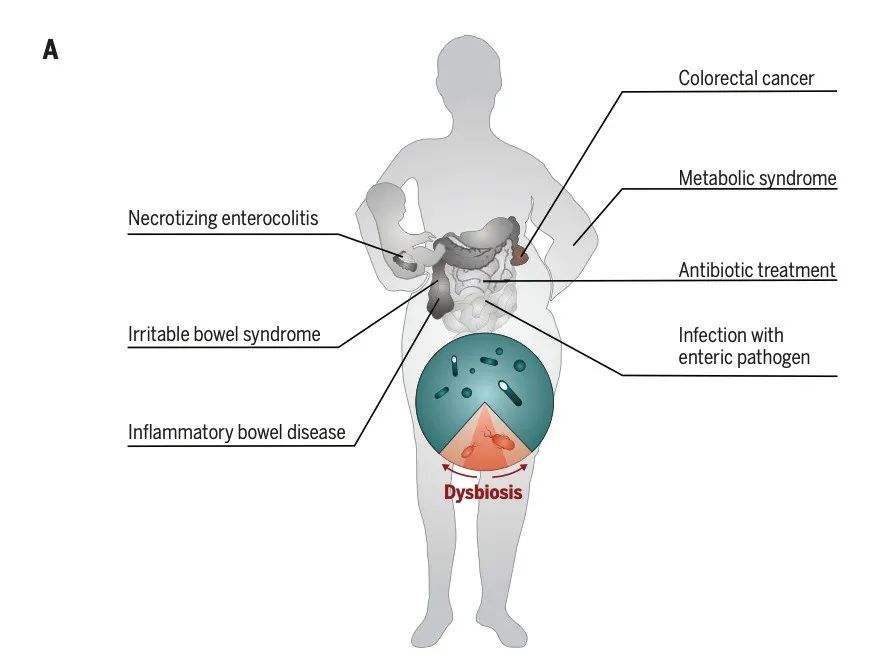
图 1A. 在健康的肠道中,专性厌氧菌(青色)是肠道微生物群中的优势菌。多种人类疾病的一个重要特征是炎症引起的肠道(菌群)失调,其特点是微生物组结构发生转变,专性厌氧菌减少、兼性厌氧菌增多,如肠杆菌科(红色)。
在健康的结肠中,肠杆菌科并非是微生物组最主要的组成部分[4]。然而,兼性厌氧的肠杆菌科的扩张往往是肠道微生物生态系统失调的特征[5](图1A)。
事实上,抗生素治疗对肠道菌群的干扰会导致肠杆菌科细菌的生长[6~10]。肠道炎症的患者的肠道菌群中出现了肠杆菌科的扩张,比如严重的肠道炎症包括炎症性肠病[11~14]、结直肠癌[15,16]、坏死性小肠结肠炎[17],以及引起低水平肠道炎症的疾病,如肠易激综合征[18,19]和肥胖[20,21]。
这些研究表明,肠杆菌科细菌的扩张可能在人类疾病的发病机制中起着重要作用,应该被认为是肠道菌群失调的微生物标志。
Mariana X. Byndloss 利用其在微生物学、免疫学和兽医学的知识背景,探究宿主和微生物组间的相互作用,以防止与疾病相关的肠杆菌科扩张[10]。
Mariana发现,栖息在结肠中的专性厌氧微生物对人体有重要的益处,它们能够将复杂的膳食碳水化合物(纤维)消化为发酵产物,从而有助于宿主营养[22]、免疫发育[23~26]和保护生态位免受病原体的伤害[10,27]。
相比之下,兼性厌氧细菌,如肠杆菌科,则不能提供这样的好处。当有氧气存在时,兼性厌氧细菌可以对上述发酵产物进行代谢,将营养物质从宿主中转移出去[28~30]。
因此,宿主很可能已经进化出了相应的策略,来维持由专性厌氧菌(可发酵纤维)主导的多样性肠道菌群,这一策略也被称为“菌群滋养性免疫(microbiota-nourishing immunity)”[31,32]。
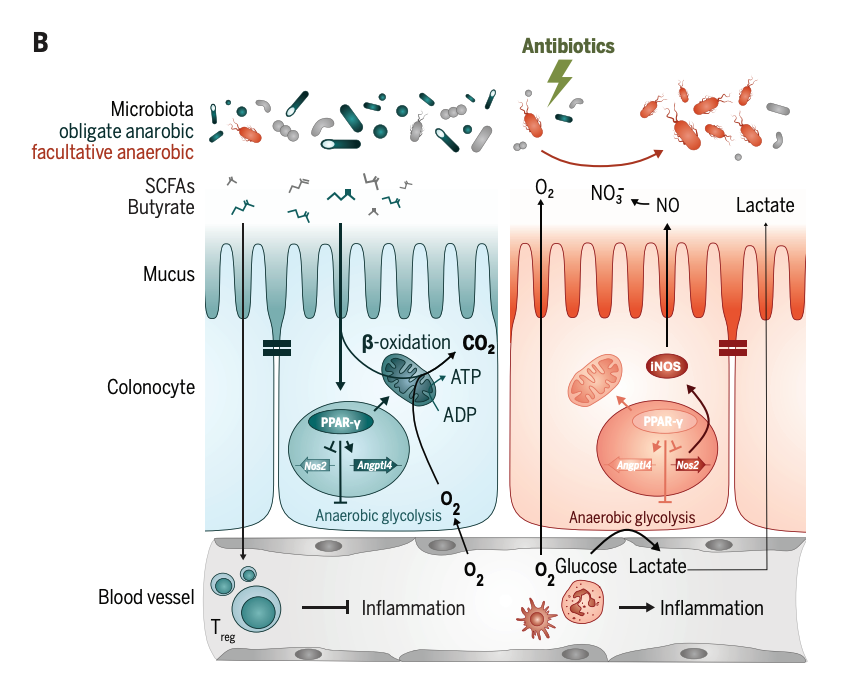
图 1B. 肠上皮功能障碍是菌群失调相关的肠杆菌科扩张的关键驱动因素
Mariana 近期的工作强调了结肠上皮细胞在形成有益微生物群和促进菌群滋养性免疫方面的中心作用(图 1B)。
结肠上皮细胞的成熟和分化需要 PPAR-γ(过氧化物酶体增殖物激活受体 γ)[33],一种在小鼠和人类分化的结肠上皮细胞中高度表达的核受体[34]。PPAR-γ 可激活线粒体对长链和短链脂肪酸的 β-氧化,通过氧化磷酸化作用消耗氧气[35~37]。
成熟的结肠上皮细胞通过消耗高水平的氧气来维持其氧化代谢状态,从而导致氧气分压低于 7.6 毫米汞柱(<1%的氧气),这种情况被称为生理性上皮缺氧[38]。
Mariana 的工作确定了:成熟结肠上皮细胞高水平的氧化代谢状态,在限制黏膜表面散发的氧气量方面起着关键作用,这有助于维持大肠腔内的厌氧环境(图 1B)[10,39]。通过这一机制,结肠上皮确保了有益厌氧微生物的优势地位,从而维持肠道稳态。
结肠上皮细胞在维持结肠稳态中的作用表明,微生物组的不平衡可能是由上皮代谢功能的潜在缺陷引起的。为了深入了解肠道内稳态破坏的机制,Mariana 采用了一个抗生素模型来实现微生物组的失调(图 1B)。
该模型通过消耗微生物来源的短链脂肪酸,如丁酸盐、丙酸盐和醋酸盐来改变上皮细胞的代谢。丁酸盐能激活人类上皮细胞中的 PPAR-γ 信号[40],以驱动黏膜表面的结肠上皮细胞中的线粒体进行 β-氧化[35~37],对维持生理性上皮缺氧至关重要[10]。
短链脂肪酸还会通过维持黏膜中调节性 T 细胞池发挥抑制肠道炎症的作用[23~26]。
Mariana 的研究证实,抗生素处理会通过下调上皮 PPAR-γ 信号和减少结肠黏膜中调节性 T 细胞的数量[10,41]导致肠道炎症的恶化[41]。
炎症信号的上调导致分化的结肠上皮细胞代谢向无氧糖酵解转变——一种以低耗氧量[10,42]为特征的代谢,会导致上皮缺氧的丧失。
上皮氧合作用增强的一个重要后果是:黏膜表面散发的氧气量增加。在该动物模型中,氧气量的增加允许兼性厌氧菌进行有氧呼吸,从而促进了肠杆菌科细菌的扩张。
PPAR-γ 能抑制促炎基因的转录,包括 iNOS 基因(Nos2)[43]。结肠上皮细胞中,菌群失调依赖性 PPAR-γ 信号的下调,导致了 iNOS 的合成增加。iNOS 是一种可以产生一氧化氮的酶,一氧化氮会在肠腔中反应形成硝酸盐,从而通过厌氧硝酸盐呼吸促进肠杆菌科细菌的扩张[10]。
Mariana 对结肠炎模型的研究揭示了肠道上皮细胞在维持肠道稳态中的作用[44]。其发现右旋糖酐硫酸钠(DSS)引起的上皮损伤会激活肠道修复反应,导致分化的结肠上皮细胞减少;黏膜表面散发的氧气量,可通过有氧呼吸促进致癌肠杆菌科细菌的生长,从而增加疾病的严重程度[44]。
综上所述,Mariana 的研究揭示了结肠上皮细胞代谢通过“窒息”有害细菌,在平衡肠道微生物组中起着核心作用[31,45]。具体地,上皮缺氧的破坏,以及随后进入结肠腔的氧气量的增加,会导致兼性厌氧菌的大量繁殖,这种细菌与肠道菌群失调和炎症性疾病有关。
此外,Mariana 的工作已经证实,结肠上皮的生理变化也会导致替代电子受体(alternative electron acceptors)水平的增加,肠杆菌科可以利用这些替代电子受体进行无氧呼吸[10]。
第三位获奖者Oliver Harrison
Oliver Harrison 获得了巴斯大学的学士学位和牛津大学的博士学位。在 NIH 完成了博士后工作后,Oliver 于 2019 年在贝纳罗亚研究所的基础免疫学中心建立了他的实验室。
他的研究探究了 T 细胞和 B 细胞对共生微生物的反应是如何促进屏障组织的完整性和修复,以及是如何导致疾病的。
以下是他的研究成果介绍:
免疫系统对健康具有强大的保护作用,当宿主面临感染或来自环境的挑战时,该系统能够保护并维持宿主的组织功能。
迄今为止,我们对于宿主免疫的理解主要来自于病原微生物感染和炎症模型。但是,绝大多数微生物与免疫间的“互动”实际上发生在共生菌群和宿主之间。共生菌群是栖息在我们机体屏障组织中(如胃肠道和皮肤)的数万亿微生物。
这些微生物从来没有被宿主忽视过,它们促进了哺乳动物免疫系统的发育、成熟1。反过来,与共生微生物相关的免疫反应,特别是那些由适应性免疫系统调节的免疫反应,能够增强上皮细胞的抗菌功能,抑制外来的致病微生物2。
存在于屏障表面的共生微生物的免疫应答具有一个突出特点,即在应答的开始和效应阶段都不会引发炎症并能维持组织的完整性。
对共生细菌的这种免疫形式引起了许多问题:特别是,对病原体的适应性免疫的常规规则在多大程度上适用于这些非侵入性微生物?以及在组织损伤期间,共生特异性 T 细胞是如何感知环境压力并产生反应的?
作为美国国立卫生研究院 Yasmine Belkaid 实验室的博士后研究员,Oliver Harrison 努力寻求解决这些问题的方法,研究了共生特异性 T 细胞反应的基本机制以及它们在组织体内平衡中的作用。
确定、追踪并描述共生特异性 T 细胞的早期工作,使 Oliver Harrison 及其同事揭示了皮肤表面的共生菌群参与宿主免疫中的三个方面:限制非经典主要组织相容性复合物(MHC)、多样的分化谱和对伤口修复的直接贡献。
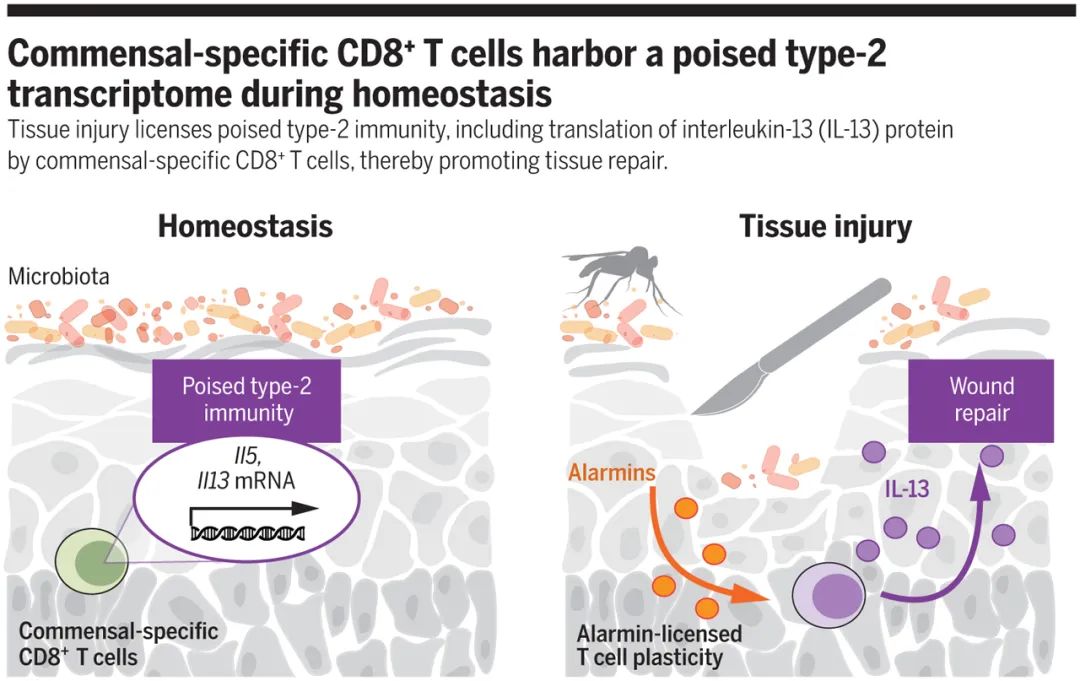
对共生菌群的非典型免疫
尽管微生物群落可能会产生大量的蛋白质,但只有少量微生物来源的抗原和抗原表位能够被共生特异性 T 细胞识别,这也为针对这些细胞的生物学机制研究造成了阻碍(3~6)。
因此,为了探索皮肤表面存在的共生特异性 T 细胞的抗原特异性,Oliver 使用了人皮肤表面常见的共生葡萄球菌的定殖新模型7。
体外筛选和计算机表位预测共同表明,共生特异性 CD8+ T 细胞应答反应与非经典 MHC1b 分子 H2-M3 协同作用,产生含有 N-甲酰甲硫氨酸(fMet)的肽8。
这些发现表明,fMet 肽,作为一种已知能够激活先天免疫系统的细菌抗原类型,也会激发对于非侵入性细菌的特异性免疫反应。同时,也强调了非经典 MHC 分子对于微生物和免疫系统之间的相互交流所具有的直接作用。
鉴定与 H2-M3 结合的 fMet 肽:建立 MHC 四聚体和 T 细胞受体转基因小鼠模型,使得他们能够在机体平衡和炎症过程中追踪并表征存在于皮肤组织内的共生特异性 CD8+ T 细胞。
利用这些工具作为体外追踪共生特异性 CD8+ T 细胞的补充方法,Oliver 发现,这些 T 细胞具有记忆特性,能够作为“哨兵”在皮肤屏障长期存活。
对于共生菌群的适度免疫
为了了解皮肤微生态是如何“培养”共生特异性 CD8+ 细胞,Oliver 等评估了它们的细胞因子和效应潜能。
共生特异性 CD8+ T 细胞与与病原体感染激活的普通 T 细胞存在明显差异,其倾向于极化成一种不同寻常的混合状态(hybrid state)。
特别是,CD8+ T 细胞表达了两种通常互斥的转录因子,即维甲酸相关的独立受体 γt(RORγt)以及 GATA 结合蛋白 3(GATA-3)。这两种转录因子对针对真菌的 17-型免疫和针对寄生虫的 2-型免疫是必不可少的。
进一步,Oliver 等通过表征这些细胞的表观遗传和转录,研究了这种共表达所造成的影响。
出乎意料的是,Oliver 等发现这些细胞会表达 2-型免疫因子 Il5 和 Il13 的 mRNA。然而,尽管这些细胞能够转录产生 Il5、Il13 的 mRNA,但是这些细胞并没有后续翻译生成出 IL-5 或 IL-13 蛋白。
因此,在健康的皮肤组织中,共生特异性 T 细胞采取了一种独特的分化模式,Oliver 将其定义为“适度的 2 型免疫(poised type-2 immunity)”9。
调节这一免疫轴的后转录过程的机制研究仍在进行之中。但是,鉴于皮肤作为保护性屏障的基础作用,他们推测适度的 2 型免疫可能受环境的影响,包括组织损伤影响。
他们猜测在局部炎症蛋白(皮肤炎症过程中释放的因子)的刺激下,受伤皮肤组织内的特异性 CD8+T 细胞会触发 2 型细胞因子蛋白质的快速翻译。
因此,2-型细胞因子的 mRNA 的表达和之后的蛋白翻译可能会与共生特异性 CD8+ T 细胞短暂地“解偶联”,使得机体能够在平衡和炎症条件下发挥不同的免疫功能。
共生特异性 T 细胞适应性可以帮助组织修复
考虑到共生特异性 CD8+ T 细胞在解剖学上接近上皮细胞以及它们在暴露于炎症介质之后显著的功能适应性,Oliver 试图了解这些皮肤上存在的这些 T 细胞对于组织修复的作用。
以创伤恢复的程度(wound bed re-epithelialization)作为组织修复的量化标志,Oliver 等观察到先前在皮肤上的共生菌群和共生体特异性 CD8+ T 细胞的聚集,都加速了伤口修复8。
皮肤上定殖的葡萄球菌对修复过程的加速作用取决于组织损伤后共生特异性 CD8+T 细胞产生 IL-13 蛋白的能力,这也反映了 MHC1b-限制性共生特异性 CD8+ T 细胞、适度 2 型免疫和组织修复之间的联系9(见图)。
借鉴共生特异性免疫的经验
通过探究共生特异性 T 细胞,Oliver 等拓展了对这种知之甚少的免疫机制的理解。其发现突显出了存在于共生菌群与免疫系统间的互作的精妙的特异性,是组织自平衡和组织修复的关键因素所在。
和其他人一样,Oliver 等猜测这种共生关系中的波动很可能会触发皮肤慢性炎症性疾病的发生和发展。更进一步了解共生特异性免疫对组织生理学的作用之后,未来可能会出现针对免疫-菌群相互作用造成的慢性炎症和疾病的个性化治疗方案。
(滑动下方查看)
第一部分:精确调节微生物组代谢
参考文献:
1. A. Almeida et al, Nature 568, 499 (2019).
2. C.-J. Guo et al., Science 366, eaav1282 (2019).
3. M. G. Rooks, W. S. Garrett, Nat. Rev. Immunol. 16, 341 (2016).
原文链接:https://science.sciencemag.org/content/369/6500/153.2
作者|Chun-Jun Guo
编译|C。
第二部分:菌群滋养性免疫
参考文献:
1. Y. Belkaid, T. W. Hand, Cell 157, 121 (2014).
2. A. M. Martin, E. W. Sun, G. B. Rogers, D. J. Keating, Front. Physiol. 10, 428 (2019).
3. B. Routy et al., Science 359, 91 (2018).
4. P. B. Eckburg et al., Science 308, 1635 (2005).
5. N. R. Shin, T. W. Whon, J.-W. Bae, Trends Biotechnol. 33, 496 (2015).
6. E. J. Vollaard, H. A. L. Clasener, A. J. H. M. Janssen, J. Antimicrob. Chemother. 30, 685 (1992).
7. S. Kieser et al., Cell. Mol. Gastroenterol. Hepatol. 5, 458 (2017).
8. M. Bohnhoff, B. L. Drake, C. P. Miller, Proc. Soc. Exp. Biol. Med. 86, 132 (1954).
9. K. Saito, Paediatr. Jpn. 65, 385 (1961).
10. M. X. Byndloss et al., Science 357, 570 (2017).
11. P. Seksik et al., Gut 52, 237 (2003).
12. U. Gophna, K. Sommerfeld, S. Gophna, W. F. Doolittle, S. J. O. Veldhuyzen van Zanten, J. Clin. Microbiol. 44, 4136 (2006).
13. X. C. Morgan et al., Genome Biol. 13, R79 (2012).
14. A. Darfeuille-Michaud et al., Gastroenterology 115, 1405 (1998).
15. H. M. Martin et al., Gastroenterology 127, 80 (2004).
16. A. Swidsinski et al., Gastroenterology 115, 281 (1998).
17. C. J. Hunter, J. S. Upperman, H. R. Ford, V. Camerini, Pediatr. Res. 63, 117 (2008).
18. L. Krogius-Kurikka et al., BMC Gastroenterol. 9, 95 (2009).
19. A. P. Gobert et al., Sci. Rep. 6, 39399 (2016).
20. N. Fei, L. Zhao, ISME J. 7, 880 (2013).
21. S. Devkota et al., Nature 487, 104 (2012).
22. O. C. Velazquez, H. M. Lederer, J. L. Rombeau, in Dietary Fiber in Health and Disease (Springer, 1997), pp. 123–134.
23. K. Atarashi et al., Science 331, 337 (2011).
24. N. Arpaia et al., Nature 504, 451 (2013).
25. Y. Furusawa et al., Nature 504, 446 (2013).
26. P. M. Smith et al., Science 341, 569 (2013).
27. F. Rivera-Chávez et al., Cell Host Microbe 19, 443 (2016).
28. L. Spiga et al., Cell Host Microbe 22, 291 (2017).
29. F. Faber et al., PLOS Pathog. 13, e1006129 (2017).
30. D. N. Bronner et al., Cell Host Microbe 23, 266 (2018).
31. M. X. Byndloss, A. J. Bäumler, Nat. Rev. Microbiol. 16, 103 (2018).
32. M. X. Byndloss, Y. Litvak, A. J. Bäumler, Inflamm. Bowel Dis. 25, 811 (2019).
33. Z. Tylichová et al., J. Nutr. Biochem. 39, 145 (2017).
34. M. Lefebvre et al., J. Endocrinol. 162, 331 (1999).
35. K. Duszka, M. Oresic, C. Le May, J. König, W. Wahli, Int. J. Mol. Sci. 18, 2559 (2017).
36. D. R. Donohoe et al., Cell Metab. 13, 517 (2011).
37. W. E. Roediger, Gut 21, 793 (1980).
38. G. T. Furuta et al., J. Exp. Med. 193, 1027 (2001).
39. Y. Litvak, M. X. Byndloss, R. M. Tsolis, A. J. Bäumler, Curr. Opin. Microbiol. 39, 1 (2017).
40. S. Alex et al., Mol. Cell. Biol. 33, 1303 (2013).
41. A. M. Spees et al., mBio 4, e00430 (2013).
42. C. C. Gillis et al., Cell Host Microbe 23, 570 (2018).
43. M. Li, G. Pascual, C. K. Glass, Mol. Cell. Biol. 20, 4699 (2000).
44. S. A. Cevallos et al., mBio 10, e02244 (2019).
45. Y. Litvak, M. X. Byndloss, A. J. Bäumler, Science 362, eaat9076 (2018).
原文链接:https://science.sciencemag.org/content/369/6500/153.1
作者|Mariana X. Byndloss
编译|赵婧
第三部分:皮肤菌群帮助伤口修复?
参考文献:
1. Y. Belkaid, O. J. Harrison, Immunity 46, 562 (2017).
2. K. Honda, D. R. Littman, Nature 535, 75 (2016).
3. Y. Cong, T. Feng, K. Fuji-hashi, T. R. Schoeb, C. O. Elson, Proc. Natl. Acad. Sci. U.S.A. 106, 19256 (2009).
4. Y. Yang et al., Nature 510, 152 (2014).
5. M. Xu et al., Nature 554, 373 (2018).
6. E. Ansaldo et al., Science 364, 1179 (2019).
7. S. Naik et al., Nature 520, 104 (2015).
8. J. L. Linehan et al., Cell 172, 784 (2018).
9. O. J. Harrison et al., Science 363, eaat6280 (2019).
原文链接:https://science.sciencemag.org/content/369/6500/152
作者|Oliver Harrison
编译|C。
审校|617